HI-VQE Chemistry - Fungsi Qiskit oleh Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Lihat rujukan API
Fungsi Qiskit adalah ciri eksperimental yang hanya tersedia untuk pengguna Pelan Premium IBM Quantum®, Pelan Flex, dan Pelan On-Prem (melalui API IBM Quantum Platform). Ia berada dalam status keluaran pratonton dan tertakluk kepada perubahan.
Package versions
Kod pada halaman ini dibangunkan menggunakan keperluan berikut. Kami mengesyorkan menggunakan versi ini atau yang lebih baharu.
qiskit-ibm-runtime~=0.45.0
Gambaran keseluruhan
Dalam kimia kuantum, masalah struktur elektronik memfokuskan pada mencari penyelesaian kepada persamaan Schrödinger elektronik — fungsi gelombang kuantum yang menggambarkan tingkah laku elektron sistem. Fungsi gelombang ini adalah vektor amplitud kompleks, dengan setiap amplitud sepadan dengan sumbangan konfigurasi elektron yang mungkin.
Keadaan dasar adalah fungsi gelombang tenaga terendah sistem dan mempunyai kepentingan istimewa dalam kajian sistem molekul. Pendekatan paling tepat untuk mengira keadaan dasar mempertimbangkan semua konfigurasi elektron yang mungkin, tetapi ini menjadi tidak dapat diuruskan untuk sistem yang lebih besar kerana bilangan konfigurasi bertumbuh secara eksponen dengan saiz sistem.
Handover Iterative Variational Quantum Eigensolver (HI-VQE) adalah kaedah hibrid kuantum-klasik yang inovatif untuk menganggar keadaan dasar sistem molekul dengan tepat. Ia mengintegrasikan perkakasan kuantum dengan pengkomputeran klasik, menggunakan pemproses kuantum untuk meneroka konfigurasi elektron calon dengan cekap dan mengira fungsi gelombang yang terhasil pada komputer klasik. Dengan menghasilkan fungsi gelombang yang padat namun tepat secara kimia, HI-VQE meningkatkan penyelidikan dan penemuan dalam kimia kuantum dan sains bahan.

HI-VQE mengurangkan kerumitan pengkomputeran masalah struktur elektronik dengan menganggar keadaan dasar secara cekap dengan ketepatan tinggi. Ia memfokuskan pada subset konfigurasi elektron yang paling relevan yang dipilih dengan teliti, mengoptimumkan ketepatan dan kecekapan.
Menggabungkan kekuatan komputer klasik dan kuantum, HI-VQE secara berulang memperhalusi dan menambah baik anggaran fungsi gelombang semasa. Teknik pembinaan subspace yang unik membantu menjadikan pemilihan konfigurasi lebih cekap, supaya pengguna mempunyai kawalan pengkomputeran yang lebih besar dan ketepatan yang lebih baik dalam simulasi kimia kuantum.
Jika anda ingin mempelajari algoritma dengan lebih mendalam, anda boleh membaca kertas penyelidikan berkaitan.
Penerangan
Bilangan konfigurasi elektron untuk sistem molekul bertumbuh secara eksponen dengan saiz sistem. Walau bagaimanapun, untuk keadaan elektronik tertentu, seperti keadaan dasar, adalah lazim bahawa hanya sebahagian kecil konfigurasi menyumbang secara signifikan kepada tenaga keadaan. Kaedah Interaksi Konfigurasi Terpilih (SCI) memanfaatkan sifat jarang ini untuk mengurangkan kos pengkomputeran dengan mengenal pasti dan memfokuskan pada konfigurasi yang paling relevan. Subset konfigurasi ini dirujuk sebagai subspace.
HI-VQE memanfaatkan kecekapan intrinsik komputer kuantum untuk mewakili sistem molekul bagi membantu pencarian subspace. Ia mengintegrasikan subrutin klasik dan kuantum untuk menyelesaikan masalah struktur elektronik dengan ketepatan tinggi. Berbeza dengan kaedah SCI kuantum sedia ada, HI-VQE menggabungkan latihan variasi, pembinaan subspace berulang, dan penapisan konfigurasi pra-pendiagonalan untuk meningkatkan kecekapan dengan mengurangkan pengukuran kuantum, lelaran, dan kos pendiagonalan klasik. Oleh itu, HI-VQE boleh diaplikasikan kepada sistem molekul yang lebih besar yang memerlukan lebih banyak qubit, dan mengurangkan kos untuk menyelesaikan masalah bersaiz tertentu kepada tahap ketepatan yang sama.
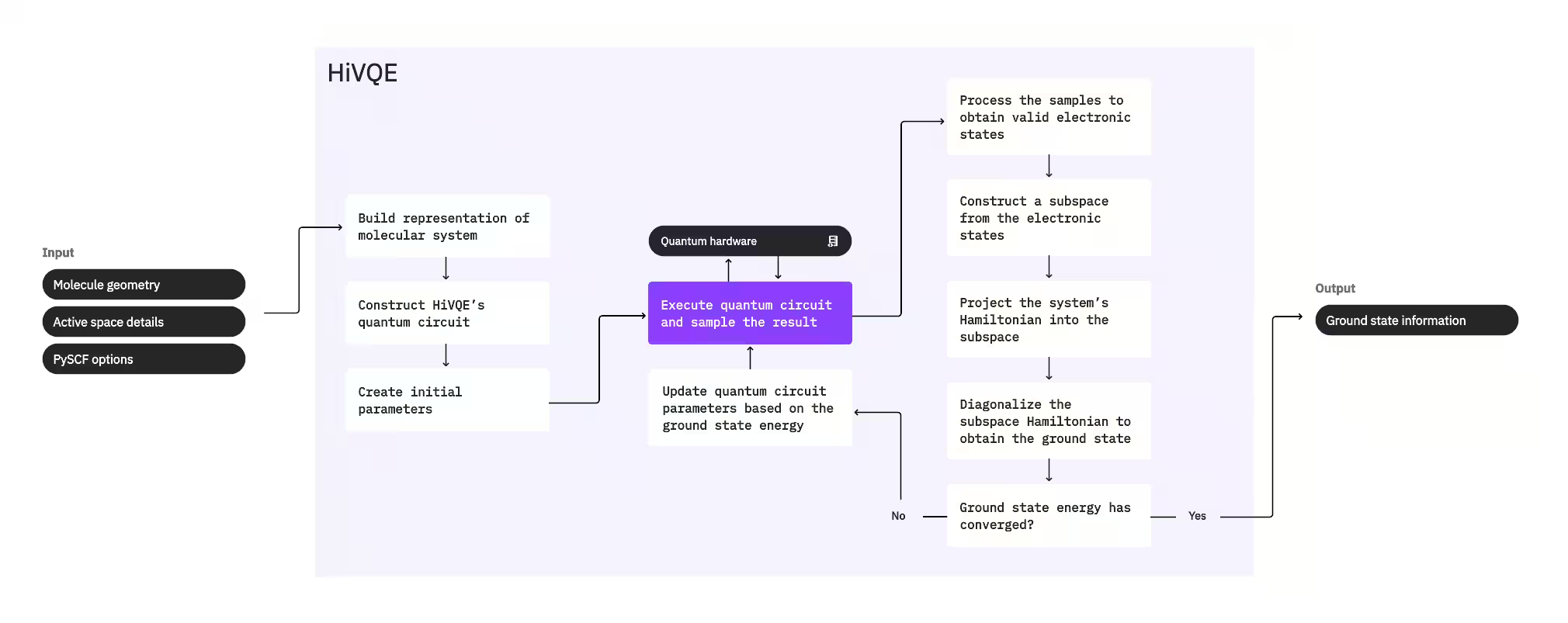
Untuk mengira keadaan dasar sistem, HI-VQE mula-mula menggunakan pakej kimia klasik PySCF untuk menjana perwakilan molekul daripada input yang diberikan pengguna, seperti geometri molekul dan maklumat molekul lain. Ia kemudian memasuki gelung pengoptimuman hibrid kuantum-klasik, secara berulang memperhalusi subspace untuk mewakili keadaan dasar secara optimum sambil meminimumkan bilangan konfigurasi yang disertakan. Gelung berterusan sehingga kriteria penumpuan, seperti saiz subspace atau kestabilan tenaga, dipenuhi, selepas itu fungsi gelombang keadaan dasar dan tenaga yang dikira dikeluarkan. Keputusan ini boleh digunakan untuk membina permukaan tenaga potensial yang tepat dan melakukan analisis lanjutan sistem.
Gelung pengoptimuman memfokuskan pada melaraskan parameter litar kuantum untuk menghasilkan subspace berkualiti tinggi. HI-VQE menawarkan tiga pilihan litar kuantum: excitation_preserving, efficient_su2, dan LUCJ. Pengoptimuman dimulakan dekat dengan keadaan rujukan Hartree-Fock kerana kesesuaiannya yang umum. litar kemudian dilaksanakan pada peranti kuantum dan konfigurasi disampling dari keadaan kuantum yang terhasil sebelum dikembalikan sebagai rentetan binari. Disebabkan hingar peranti kuantum, beberapa konfigurasi yang disampling mungkin tidak sah secara fizikal, gagal memulihara bilangan elektron atau spin. HI-VQE menangani ini menggunakan proses pemulihan konfigurasi dari pakej qiskit-addon-sqd, supaya pengguna boleh sama ada membetulkan konfigurasi tidak sah atau membuangnya.
Konfigurasi yang sah kemudian melalui langkah penapisan pilihan untuk mengalih keluar yang dijangka menyumbang secara minimal. Ini mengurangkan dimensi subspace, seterusnya menurunkan kos langkah pendiagonalan. Jika penapisan diaktifkan, Hamiltonian subspace awal dibina dari konfigurasi yang sah dan pendiagonalan dilakukan dengan kriteria penamatan yang sangat longgar. Walaupun ketepatan amplitud yang terhasil untuk setiap konfigurasi adalah rendah, ia berkesan untuk meramalkan konfigurasi mana yang hendak ditinggalkan daripada subspace pada lelaran ini, dan ia cepat untuk dikira.
Konfigurasi yang dipilih ditambahkan ke subspace, dan Hamiltonian sistem diprojeksikan ke dalam subspace ini. Subspace dikemas kini secara berulang, mengekalkan konfigurasi yang paling relevan merentasi lelaran. Pendekatan ini berbeza dengan kaedah alternatif kerana litar kuantum tidak perlu menghampiri keadaan dasar penuh pada setiap langkah.
Seterusnya, Hamiltonian subspace didiagonalkan secara klasik untuk mendapatkan nilai eigen terendah dan eigenvector yang sepadan, mewakili penghampiran keadaan dasar dan tenaganya. Apabila kualiti subspace bertambah baik melalui lelaran, keadaan dasar yang dikira lebih menghampiri keadaan dasar sebenar. Langkah penapisan tambahan boleh dilakukan pada ketika ini untuk mengalih keluar konfigurasi daripada subspace yang tidak mempunyai sumbangan ketara kepada keadaan dasar yang dikira. Langkah ini memastikan subspace yang dibawa ke lelaran seterusnya adalah sepadat mungkin. Ini dinilai berdasarkan amplitud yang dikembalikan oleh pendiagonalan, kerana ini mewakili kepentingan sumbangan setiap konfigurasi kepada keadaan dasar yang dikira.
Semakan penumpuan kemudian menentukan sama ada latihan lanjut akan meningkatkan keputusan. Jika ya, langkah pengembangan klasik pilihan dilakukan, parameter litar kuantum dikemas kini untuk lebih meminimumkan tenaga yang dikira, dan proses berulang. Langkah pengembangan klasik menjana konfigurasi tambahan untuk subspace, melengkapi konfigurasi yang disampling dari peranti kuantum. Ia mula-mula mengenal pasti konfigurasi dengan amplitud terbesar dalam keputusan pendiagonalan, sebelum menjana konfigurasi baharu dengan eksitasi tunggal dan berganda dari konfigurasi yang dikenal pasti. Bilangan konfigurasi yang dikehendaki kemudian ditambahkan ke subspace.
Setelah ditentukan bahawa lelaran telah menumpu, HI-VQE mengembalikan keadaan dasar yang dikira (dalam bentuk keadaan dalam subspace dan amplitud mereka dalam fungsi gelombang keadaan dasar), tenaganya, dan ukuran varians tenaga yang memberi petunjuk sama ada keadaan yang dikira membentuk eigenstate Hamiltonian sistem.
Pengguna boleh menentukan litar kuantum yang digunakan dan bilangan shot yang diambil untuk setiap litar kuantum, serta mengawal saiz subspace atau membolehkan penjanaan klasik konfigurasi tambahan untuk membantu konfigurasi yang dijana kuantum. Dengan cara ini pengguna boleh menyesuaikan tingkah laku HI-VQE untuk memenuhi aplikasi yang dikehendaki.
Pelesenan
Sila ambil perhatian bahawa penggunaan Fungsi Qiskit ini terhad kepada masalah yang memerlukan paling banyak 20 qubit, melainkan lesen diperoleh yang memberikan had yang lebih tinggi.
Sila e-mel qiskit.support@qunovacomputing.com jika anda ingin bertanya tentang memperoleh lesen.
Mulakan
Pertama, mohon akses ke fungsi. Kemudian, sahkan menggunakan kunci API IBM Quantum® anda dan, dengan mengandaikan anda telah menyimpan akaun anda ke persekitaran tempatan anda, pilih Fungsi Qiskit seperti berikut:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Contoh
Contoh pertama menunjukkan cara mengira tenaga keadaan dasar untuk molekul NH3 menggunakan algoritma HI-VQE.
Takrifkan geometri molekul dan pilihan
Geometri molekul NH3 diberikan dengan koordinat Kartesian yang dipisahkan dengan ";" untuk setiap atom.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Pilihan tambahan boleh ditakrifkan dan diberikan untuk sistem molekul dalam format kamus berikut.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Laksanakan fungsi dengan input geometri dan pilihan.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Adalah baik untuk mencetak ID kerja Fungsi supaya ia boleh diberikan dalam permintaan sokongan jika ada yang tidak kena.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Contoh ini kemudian menggunakan 16 qubit dengan 8 orbital asas sto3g untuk molekul NH3. Semak status atau kembalikan keputusan beban kerja Fungsi Qiskit anda seperti berikut:
print(job.status())
QUEUED
Selepas kerja selesai, keputusan boleh diperoleh dengan instans result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Untuk mengakses tenaga keadaan dasar, gunakan kunci "energy". Kunci "eigenvector" menyediakan pekali CI dengan notasi bitstring konfigurasi elektron yang sepadan yang disimpan dengan "states" dalam keputusan.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Prestasi
Bahagian ini menunjukkan pengiraan penanda aras yang ditunjukkan HI-VQE dengan kes 24-qubit untuk Li2S, kes 40-qubit untuk molekul N2, dan kes 44-qubit untuk sistem FeP-NO.
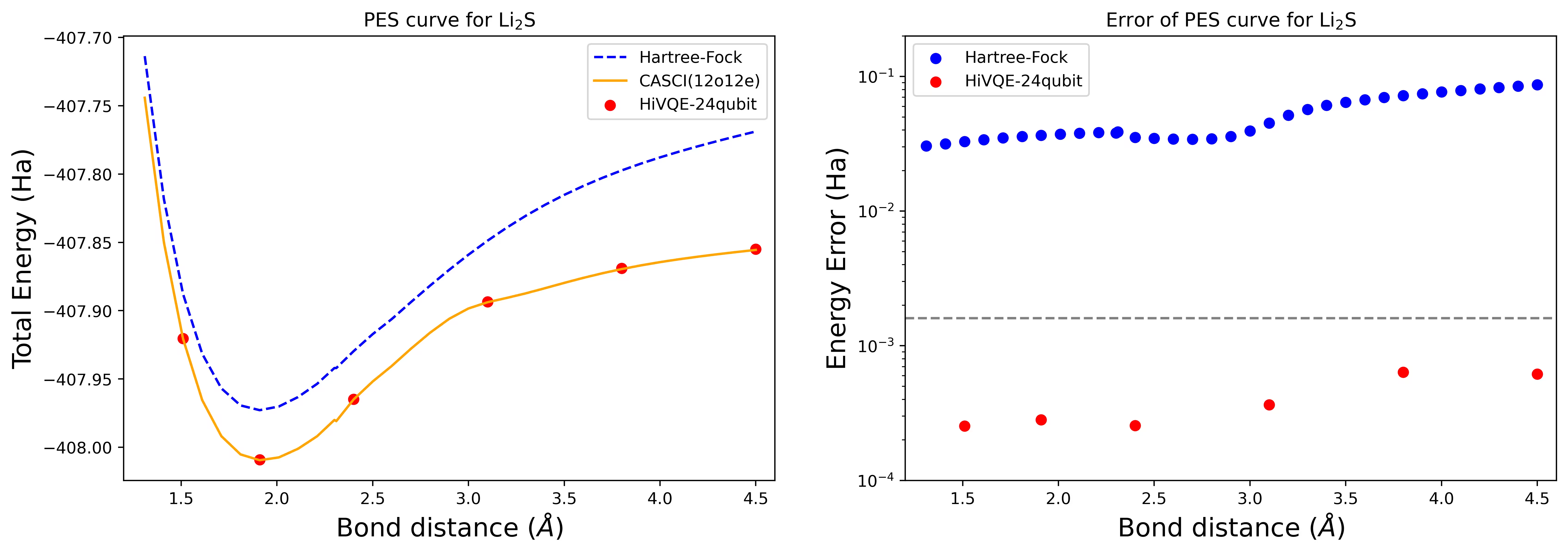
Lengkung permukaan tenaga potensial dissosiasi untuk molekul Li2S dengan 24 qubit
Lengkung PES ditunjukkan dengan rujukan FCI dan anggaran awal dari RHF, bersama-sama dengan ralat tenaga dari rujukan FCI.
Pengiraan telah dijalankan dengan geometri dan pilihan berikut.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Titik merah mewakili keputusan pengiraan HI-VQE untuk enam geometri berbeza, dan tiga geometri yang sepadan dengan 1.51, 2.40, dan 3.80 Angstrom disediakan sebagai input dalam sel di atas.
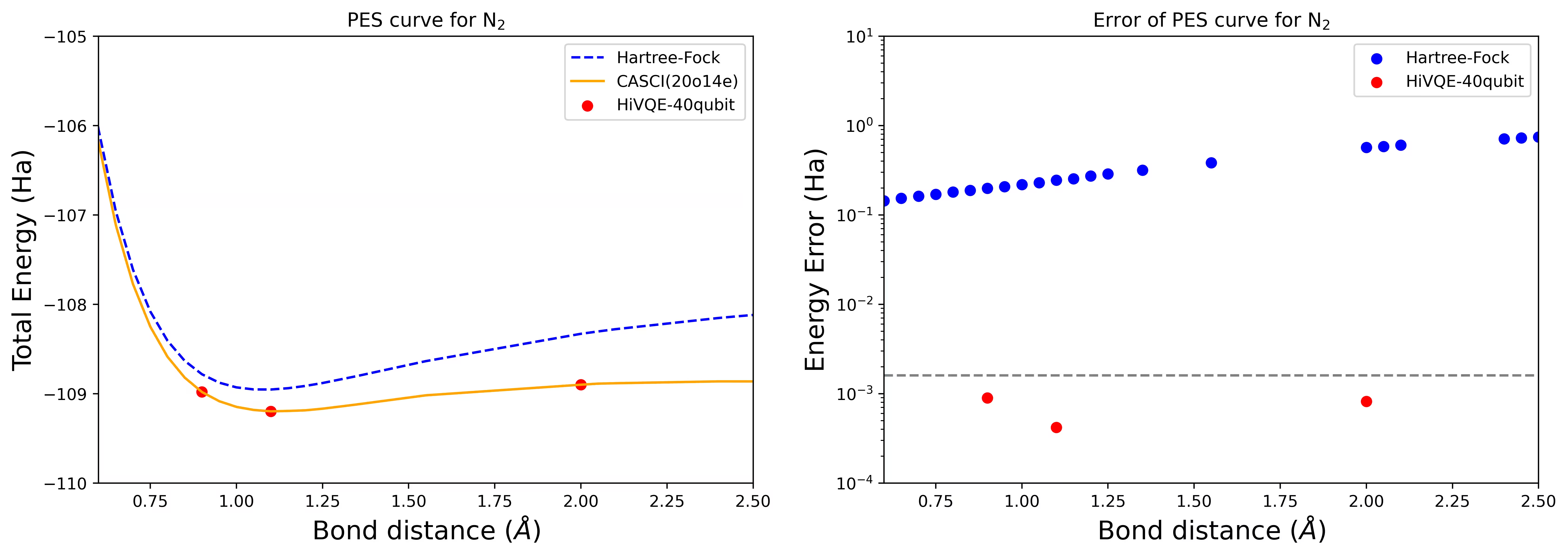
Lengkung PES dissosiasi untuk molekul N2 dengan 40 qubit
Molekul nitrogen telah dikenal pasti sebagai sistem multirujukan dengan sumbangan tenaga korelasi yang besar melebihi keadaan Hartree-Fock. Kami menjalankan pengiraan penanda aras untuk molekul N2 dengan asas cc-pvdz, (20o,14e) menggunakan pemilihan orbital aktif homo-lumo. Nombor ruang aktif penuh (CAS) untuk mewakili masalah ini ialah 6,009,350,400. Tidak mungkin mendapatkan penyelesaian masalah nilai eigen (untuk tenaga dan struktur elektronik) dengan bilangan keadaan ini menggunakan komputer meja yang berkuasa (16cpu/64GB). Dengan HI-VQE, pengguna boleh mencari subspace keadaan CAS dengan cekap untuk mencari keputusan yang tepat secara kimia sambil menjimatkan sumber pengkomputeran dengan ketara. Plot berikut menunjukkan lengkung PES pengiraan HI-VQE 40 qubit untuk dissosiasi molekul N2.
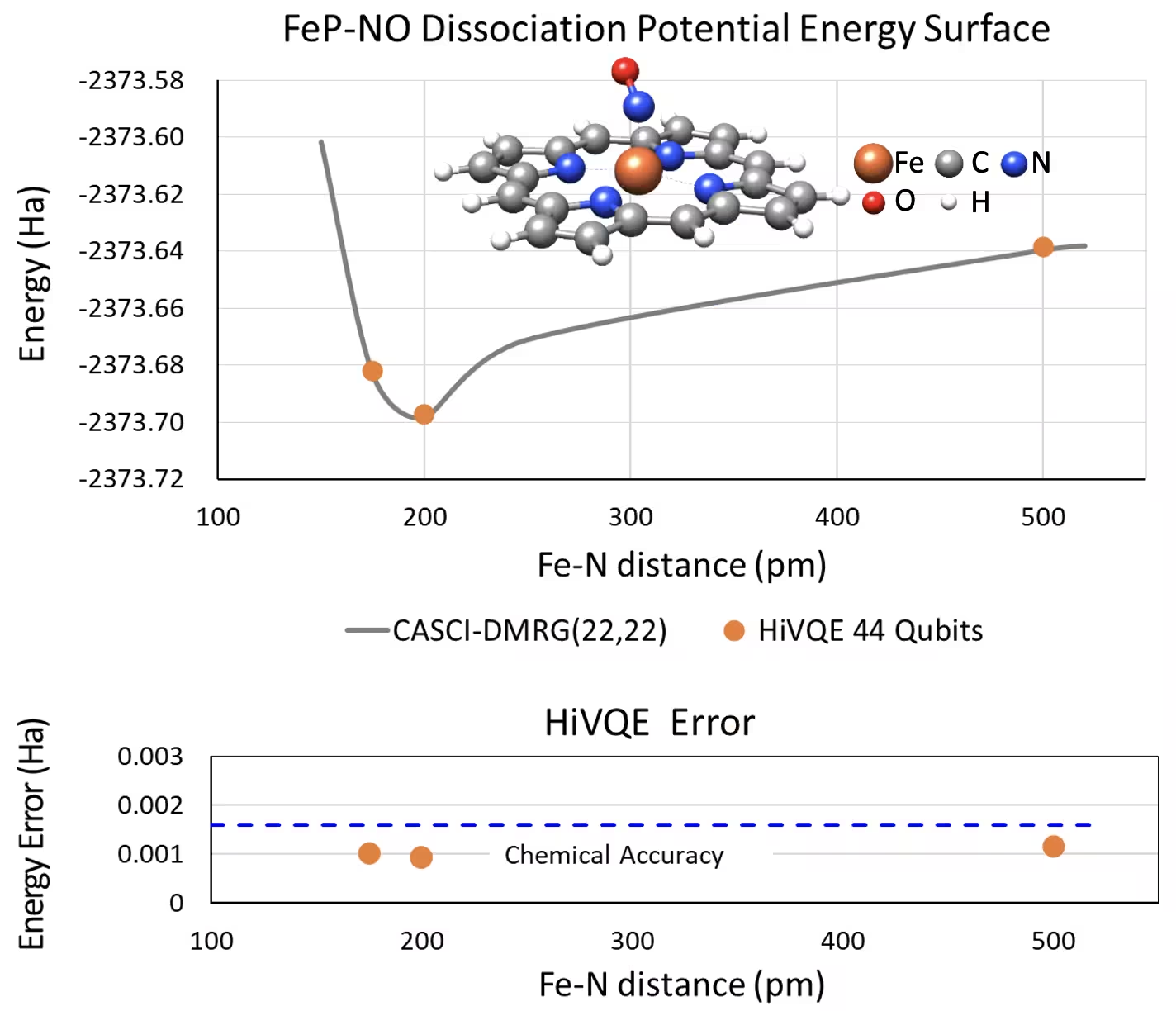
Lengkung PES dissosiasi untuk iron(II)-porfirin berlima koordinat dengan sistem NO dengan 44 qubit
Sistem kimia menarik lain ialah kompleks iron(II)-porfirin (FeP) dengan ligan nitrik oksida (NO) terkoordinat, yang mewakili sistem metaloporfirin yang berkaitan biologi yang memainkan peranan penting dalam pelbagai proses fisiologi. Dalam contoh ini, HI-VQE telah digunakan untuk menganggar lengkung permukaan tenaga potensial yang tepat bagi interaksi antarmolekul antara FeP dan NO (tenaga keadaan dasar untuk geometri yang dipisahkan secara berbeza). Sistem gabungan mempunyai 450 orbital dan 202 elektron (450o,202e) dengan asas 6-31g(d) secara keseluruhan. Pemilihan orbital aktif homo-lumo digunakan untuk mengira kes yang lebih kecil dari kes sebenar dengan (22o,22e). Daripada keputusan penanda aras berikut, kami berjaya mencapai ketepatan kimia (> 1.6 mHa) dengan pengiraan kimia komputer klasik terkini CASCI(DMRG) (22o,22e) sebagai rujukan.
Penanda aras
- Saiz matriks tepat adalah bilangan determinan untuk penyelesaian tepat, seperti FCI dan CASCI.
- Pengiraan HI-VQE mengambil sampel dan mengira subspace daripadanya (iaitu, saiz matriks HI-VQE).
- Jumlah masa termasuk masa jalan QPU dan jalankan Fungsi Qiskit dengan CPU.
- Ketepatan dianggar daripada perbezaan tenaga dari penyelesaian tepat.
| Sistem kimia | Bilangan qubit | Saiz matriks tepat | Saiz matriks HI-VQE | E(perbezaan) dari tepat (mHa) | Bilangan lelaran | Jumlah masa | Penggunaan masa jalan QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
Dapatkan mesej ralat
Jika beban kerja anda gagal, status akan menjadi ERROR dan memanggil job.result() akan menaikkan pengecualian:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Dapatkan sokongan
Anda boleh menghantar e-mel ke qiskit.support@qunovacomputing.com untuk bantuan dengan fungsi ini.
Jika anda memerlukan bantuan untuk menyelesaikan masalah ralat tertentu, sila berikan ID kerja Fungsi bagi kerja yang menghadapi ralat tersebut.
Langkah seterusnya
- Mohon akses ke fungsi dengan mengisi borang ini.
- Lawati rujukan API untuk Fungsi Qiskit ini.
- Cuba tutorial Kira lengkung PES dissosiasi untuk FeP-NO dengan HI-VQE.
- Semak Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Cuba tutorial Lengkung PES dissosiasi dengan Qunova HiVQE.