Variational Quantum Eigensolver (VQE)
Untuk modul ini, pelajar perlu mempunyai persekitaran Python yang berfungsi, serta versi terkini pakej-pakej berikut dipasang:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Untuk menyediakan dan memasang pakej-pakej ini, rujuk panduan Pasang Qiskit. Untuk menjalankan kerja pada komputer kuantum sebenar, pelajar perlu menyediakan akaun IBM Cloud, mengikuti langkah-langkah dalam panduan Sediakan akaun IBM Cloud anda.
Modul ini telah diuji dan menggunakan lebih kurang 8 minit masa QPU. Ini adalah anggaran, dan penggunaan sebenar anda mungkin berbeza.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Pengenalan
Sejak pembangunan model mekanik kuantum pada awal abad ke-20, saintis telah memahami bahawa elektron tidak mengikuti laluan tetap mengelilingi nukleus atom, sebaliknya wujud dalam kawasan kebarangkalian yang dipanggil orbital. Orbital-orbital ini sepadan dengan aras tenaga diskrit tertentu yang boleh diduduki oleh elektron. Elektron secara semula jadi berada pada aras tenaga terendah yang tersedia, yang dikenali sebagai keadaan asas. Walau bagaimanapun, jika elektron menyerap tenaga yang mencukupi, ia boleh melompat ke aras tenaga yang lebih tinggi, memasuki keadaan teruja. Keadaan teruja ini bersifat sementara, dan elektron akhirnya akan kembali ke aras tenaga yang lebih rendah, melepaskan tenaga yang diserap, selalunya dalam bentuk cahaya. Proses asas penyerapan dan pelepasan tenaga ini penting untuk memahami cara atom berinteraksi dan membentuk ikatan.
Apabila atom-atom bergabung membentuk molekul, orbital atom mereka bergabung membentuk orbital molekul. Susunan dan aras tenaga elektron dalam orbital molekul ini menentukan sifat molekul yang terhasil dan kekuatan ikatan kimia. Contohnya, dalam pembentukan molekul hidrogen () daripada dua atom hidrogen individu, elektron daripada setiap atom menduduki orbital atom. Apabila atom-atom saling menghampiri, orbital atom ini bertindih dan bergabung membentuk orbital molekul baru — satu dengan tenaga lebih rendah (orbital ikatan) dan satu dengan tenaga lebih tinggi (orbital anti-ikatan). Kedua-dua elektron, satu dari setiap atom hidrogen, akan lebih suka menduduki orbital ikatan bertenaga rendah, yang membawa kepada pembentukan ikatan kovalen stabil yang menyatukan molekul . Perbezaan tenaga antara atom-atom yang terpisah dan molekul yang terbentuk, khususnya tenaga elektron dalam orbital molekul, menentukan kestabilan dan sifat-sifat ikatan.
Dalam bahagian-bahagian berikut, kita akan meneroka proses pembentukan molekul ini, dengan menumpukan perhatian pada molekul . Kita akan menggunakan komputer kuantum sebenar, digabungkan dengan teknik pengoptimuman klasik, untuk mencari tenaga proses asas namun penting ini. Eksperimen ini akan memberikan demonstrasi praktikal tentang bagaimana pengiraan kuantum boleh digunakan untuk menyelesaikan masalah dalam kimia komputasi, memberikan gambaran tentang peranan tenaga elektron.
VQE - Algoritma kuantum variatif untuk masalah nilai eigen
Teknik penghampiran untuk kimia - prinsip variatif dan set asas
Sumbangan Erwin Schrödinger kepada mekanik kuantum tidak terhad kepada memperkenalkan model elektronik baru; secara asasnya, beliau menetapkan mekanik gelombang dengan membangunkan persamaan Schrödinger bersandar masa yang terkenal:
Di sini, ialah operator Hamiltonian, yang mewakili jumlah tenaga sistem, dan ialah fungsi gelombang yang mengandungi semua maklumat tentang keadaan kuantum sistem. (Nota: ialah terbitan masa penuh, dan kita tidak menyertakan nilai eigen tenaga secara eksplisit di sini.)
Walau bagaimanapun, dalam banyak aplikasi praktikal — seperti menentukan aras tenaga yang dibenarkan bagi atom dan molekul — kita sebaliknya menggunakan persamaan Schrödinger tak bersandar masa (persamaan nilai eigen tenaga), yang diterbitkan daripada bentuk bersandar masa dengan menganggap keadaan pegun. Keadaan pegun ialah keadaan kuantum di mana ketumpatan kebarangkalian mencari zarah pada titik tertentu dalam ruang tidak berubah mengikut masa.
Dalam bentuk ini, mewakili nilai eigen tenaga yang sepadan dengan keadaan kuantum . Hamiltonian merangkumi pelbagai sumbangan tenaga, seperti tenaga kinetik elektron dan nukleus, daya tarikan antara elektron dan nukleus, dan daya tolakan antara elektron.
Menyelesaikan persamaan nilai eigen tenaga membolehkan kita mengira aras tenaga terkuantum bagi sistem atom dan molekul. Walau bagaimanapun, untuk molekul, menyelesaikannya secara tepat adalah sukar kerana fungsi gelombang , yang menggambarkan taburan spatial elektron, adalah kompleks dan berdimensi tinggi.
Akibatnya, saintis menggunakan teknik penghampiran untuk mendapatkan penyelesaian yang praktikal dan tepat. Dalam kerja ini, kita akan menumpukan pada dua kaedah utama:
-
Prinsip variatif
Kaedah ini menghampiri fungsi gelombang dan menyesuaikannya untuk mendapatkan nilai sedekat mungkin dengan tenaga sasaran, biasanya tenaga keadaan asas sistem. Idea utama di sebalik prinsip variatif adalah mudah:
- Jika kita meneka fungsi gelombang (sebuah "fungsi percubaan"), tenaga yang dikira daripadanya akan sentiasa sama dengan atau lebih tinggi daripada tenaga keadaan asas () sistem.
- Dengan menyesuaikan parameter dalam fungsi percubaan, , kita boleh mendapatkan penghampiran tenaga keadaan asas yang semakin baik.
- Ketepatannya sangat bergantung pada pilihan fungsi gelombang percubaan . Fungsi percubaan yang dipilih dengan buruk mungkin menghasilkan anggaran tenaga yang jauh dari tepat.
-
Penghampiran set asas
Kaedah penghampiran kedua hadir pada peringkat pembinaan fungsi gelombang — pendekatan set asas. Dalam kimia kuantum, menyelesaikan persamaan Schrödinger secara tepat untuk molekul adalah hampir mustahil. Sebaliknya, kita menghampiri fungsi gelombang berbilang elektron yang kompleks dengan membinanya daripada fungsi matematik yang lebih mudah dan telah ditetapkan. Set asas pada asasnya ialah koleksi fungsi matematik yang diketahui ini, biasanya berpusat pada atom-atom dalam molekul, yang digunakan sebagai blok binaan untuk mewakili bentuk dan tingkah laku elektron dalam sistem. Bayangkan seperti cuba mencipta semula arca yang terperinci menggunakan hanya koleksi bata LEGO piawai — semakin banyak jenis dan saiz bata yang anda ada (semakin besar set asas), semakin tepat anda boleh menghampiri bentuk asal.
Fungsi asas ini sering terinspirasi daripada penyelesaian analitik untuk sistem mudah seperti atom hidrogen, mengambil bentuk seperti fungsi Gaussian atau jenis Slater, walaupun ia masih merupakan penghampiran. Daripada bekerja dengan orbital molekul penuh yang secara teori "tepat" tetapi tidak boleh dikendalikan, kita menyatakannya sebagai gabungan linear (jumlah dengan pekali) fungsi-fungsi asas ini. Kaedah ini dikenali sebagai pendekatan Gabungan Linear Orbital Atom (LCAO) apabila fungsi asas menyerupai orbital atom. Dengan mengoptimumkan pekali dalam gabungan linear ini, kita boleh mencari fungsi gelombang anggaran terbaik dan tenaga dalam had set asas yang dipilih.
- Semakin banyak fungsi yang dimasukkan dalam set asas, semakin baik penghampiran, tetapi ini datang dengan kos usaha pengiraan yang lebih tinggi.
- Set asas yang kecil memberikan anggaran kasar, manakala set asas yang besar memberikan keputusan yang lebih tepat dengan kos keperluan sumber pengiraan yang lebih banyak.
Ringkasnya, untuk menjadikan pengiraan boleh dilaksanakan dan mengurangkan kos pengiraan, kita menggunakan prinsip variatif dengan menghampiri fungsi gelombang, yang mengurangkan kerumitan pengiraan dan membolehkan pengoptimuman berulang untuk meminimumkan tenaga. Sementara itu, pendekatan set asas memudahkan pengiraan dengan mewakili orbital atom sebagai gabungan fungsi yang telah ditetapkan, bukannya menyelesaikan fungsi gelombang berterusan secara langsung.
Semak kefahaman anda
Pertimbangkan fungsi gelombang percubaan di mana ialah pemalar normalisasi dan ialah parameter boleh laras.
(a) Normalkan fungsi gelombang percubaan dengan menentukan supaya .
Jawapan
Untuk menormalkan fungsi gelombang percubaan yang diberikan:
Gunakan kamiran Gaussian:
tetapkan kemudian dapatkan:
(b) Kira nilai jangkaan Hamiltonian yang diberikan oleh di mana , yang sepadan dengan potensi pengayun harmonik mudah.
Jawapan
Hamiltonian untuk pengayun harmonik ialah:
Nilai jangkaan tenaga kinetik
Mengambil terbitan kedua:
Maka:
Menggunakan keputusan kamiran Gaussian piawai:
Nilai jangkaan tenaga keupayaan
Menggunakan:
kita dapatkan:
Nilai jangkaan jumlah tenaga
(c) Gunakan prinsip variatif untuk mencari optimum dengan meminimumkan .
Jawapan
Optimumkan untuk tenaga minimum
Bezakan
Menyelesaikan:
Menggantikan ke dalam :
yang sepadan dengan tenaga keadaan asas pengayun harmonik kuantum tepat.
VQE (Variational Quantum Eigensolver)
Variational quantum eigensolver (VQE) ialah kaedah utama yang akan kita gunakan untuk meneroka proses , dan di sini, kita akan melihat apa itu VQE dan bagaimana ia berfungsi. Tetapi mari kita berhenti sejenak dan lihat satu perkara yang sangat penting melalui soalan daftar masuk.
Semak kefahaman anda
Jika kita sudah mempunyai begitu banyak strategi untuk masalah kimia, kenapa kita perlukan komputer kuantum? Dan apakah tujuan menggunakan komputer kuantum dan klasik bersama-sama?
Jawapan
Pengkomputeran kuantum berpotensi merevolusikan kimia dengan menangani masalah yang sukar diselesaikan oleh komputer klasik akibat penskalaan eksponen keadaan kuantum. Richard Feynman terkenal mencatatkan bahawa untuk mensimulasikan alam semula jadi, pengiraan juga mesti bersifat kuantum [ref 1].
Contohnya, mensimulasikan kafein dengan set asas paling mudah (STO-3G) memerlukan bit, jauh lebih besar daripada jumlah keseluruhan bintang dalam alam semesta yang boleh diperhatikan () [ref 2]. Komputer kuantum boleh menggambarkan orbital elektronik kafein dengan 160 qubit.
Komputer kuantum secara semula jadi memproses interaksi kuantum menggunakan superposisi dan jalinan, yang menyediakan cara menjanjikan untuk membolehkan simulasi molekul yang tepat. Selanjutnya, kita boleh menggabungkan kelebihan komputer kuantum (simulasi elektron) dan komputer klasik (pra/pasca-pemprosesan data, pengurusan proses algoritma, pengoptimuman, dan sebagainya). Ini dijangka dapat meningkatkan penemuan bahan, reka bentuk ubat, dan ramalan tindak balas, mengurangkan eksperimen cuba jaya yang mahal. [ref 3][ref 4]
Jika anda ingin tahu mengapa komputer kuantum diperlukan untuk masalah kimia dan mengapa menggunakan sumber pengkomputeran kuantum dan klasik bersama, semak artikel-artikel berikut:
Sekarang mari kita kembali kepada VQE.
VQE menggabungkan kuasa komputer kuantum dengan komputer klasik, pada asasnya menggunakan prinsip variatif untuk mendapatkan tenaga keadaan asas sistem. Untuk memahami VQE, bahagikannya dahulu kepada tiga bahagian:
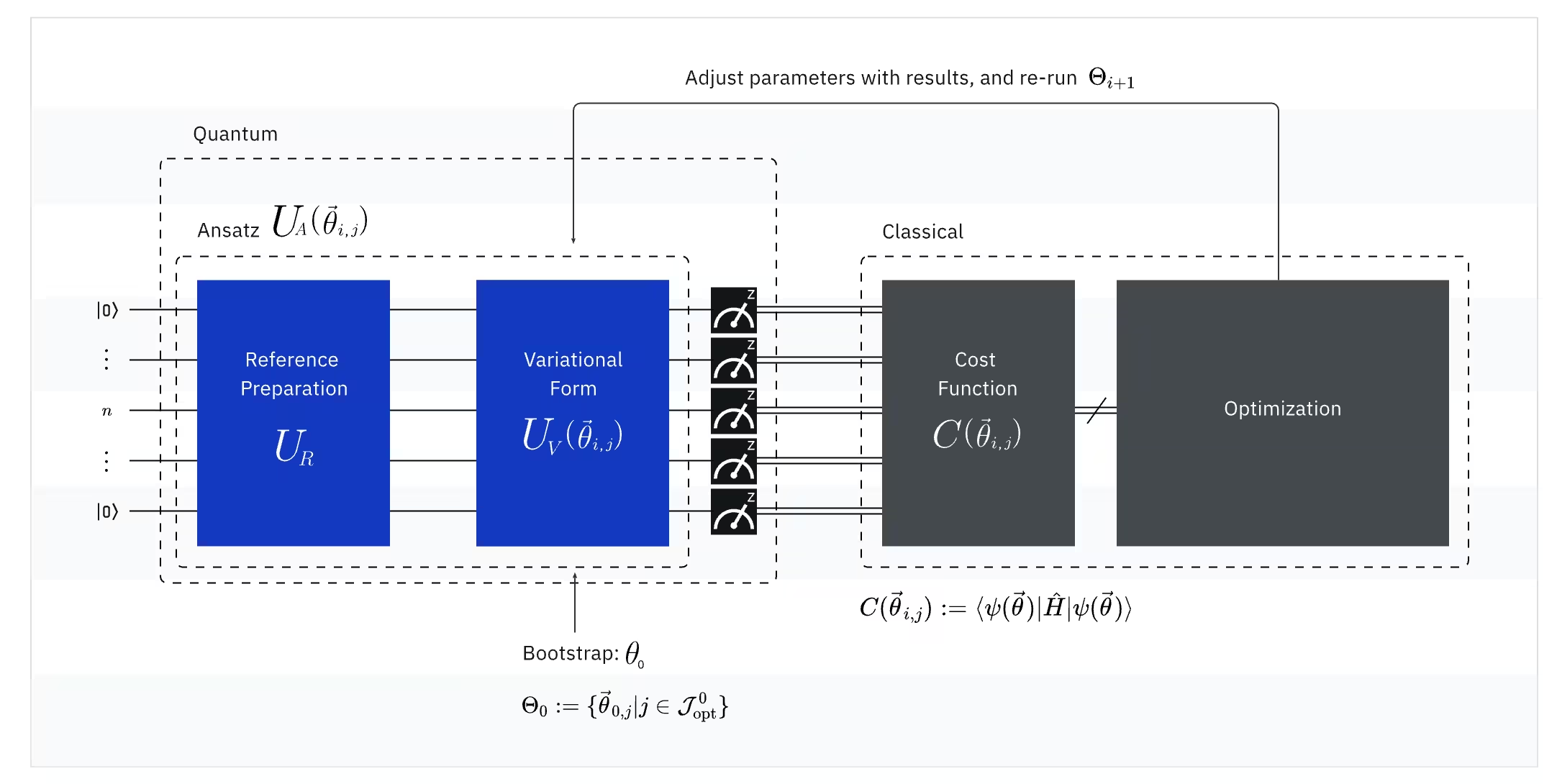
(Kuantum) Observable: Hamiltonian molekul (tenaga molekul)
Dalam VQE, Hamiltonian molekul/atom ialah satu observable, bermakna kita boleh mengukur nilainya melalui eksperimen. Matlamat kita adalah untuk mencari tenaga terendah yang mungkin (tenaga keadaan asas) bagi molekul. Untuk melakukan ini, kita menggunakan keadaan kuantum percubaan, yang dijana oleh litar kuantum berparameter (ansatz). Kita mengukur observable dan mengoptimumkan keadaan kuantum sehingga mencapai tenaga terendah yang mungkin.
Set asas yang digunakan untuk Hamiltonian molekul menentukan bilangan qubit yang diperlukan dan secara langsung mempengaruhi ketepatan VQE. Memilih set asas yang betul adalah kritikal untuk mengimbangi kecekapan dan ketepatan. Untuk memudahkan pengiraan tanpa mengubah set asas, kita boleh menggunakan strategi seperti pengenaan simetri dan pengurangan ruang aktif. Banyak molekul mempunyai bentuk simetri (seperti rama-rama atau kepingan salji), bermakna beberapa bahagian berkelakuan sama. Daripada mengira semuanya secara berasingan, kita boleh menumpukan hanya pada bahagian unik, menjimatkan sumber kuantum, dengan itu memanfaatkan simetri. Dalam pengurangan ruang aktif, kita hanya mempertimbangkan orbital yang penting, kerana tidak semua elektron memberi kesan ketara kepada tenaga molekul. Elektron yang dekat dengan nukleus kekal hampir tidak berubah, manakala yang lain mempengaruhi ikatan. Dengan menggunakan kaedah-kaedah ini, kita boleh menjadikan VQE lebih cekap sambil mengekalkan ketepatan.
Setelah kita mendapatkan Hamiltonian molekul menggunakan set asas dan strategi yang betul di atas, kita perlu mengubah Hamiltonian ini menjadi satu yang sesuai untuk komputer kuantum. Memetakan masalah kepada operator Pauli boleh menjadi cukup rumit. Ini terutamanya benar dalam kimia kuantum, yang bekerja dengan zarah yang tidak dapat dibezakan (elektron), kerana qubit adalah boleh dibezakan. Kita tidak akan masuk ke dalam butiran pemetaan di sini, tetapi kami merujuk anda kepada sumber-sumber berikut. Perbincangan umum tentang memetakan masalah kepada operator kuantum boleh didapati dalam Pengkomputeran kuantum dalam amalan. Perbincangan lebih terperinci tentang memetakan masalah kimia kepada operator kuantum boleh didapati dalam Kimia kuantum dengan VQE.
Untuk modul ini, kami akan menyediakan Hamiltonian (satu-qubit) yang sesuai untuk dan supaya kita boleh menumpukan pada penggunaan komputer kuantum. Hamiltonian satu-qubit ini disediakan dengan menggunakan set asas STO-6G dan pemetaan Jordan-Wigner, yang merupakan pemetaan paling mudah dengan interpretasi fizikal yang paling ringkas, kerana ia memetakan penghunian satu spin-orbital kepada penghunian satu qubit. Juga, kami menggunakan teknik pengurangan qubit dengan menggunakan simetri Hamiltonian, yang menggunakan corak dalam cara penghunian spin berkelakuan untuk mengurangkan bilangan qubit. Untuk molekul , kami menganggap jarak antara dua atom hidrogen ialah 0.735 .
(Kuantum) Ansatz: Fungsi gelombang percubaan (Cara membina keadaan kuantum percubaan dengan litar kuantum)
Untuk VQE, ansatz (jamak: ansätze) terdiri daripada dua komponen utama. Yang pertama ialah penyediaan keadaan awal, yang menetapkan keadaan qubit dengan menggunakan get kuantum tanpa parameter variatif. Komponen kedua ialah litar kuantum berparameter, sebuah litar kuantum khas dengan parameter boleh laras, sama seperti tombol pada radio. Parameter-parameter ini akan digunakan untuk bahagian terakhir — pengoptimum klasik — untuk membantu kita mencapai keadaan asas terbaik yang mungkin.
Dalam bahagian prinsip variatif, kita telah belajar bahawa kualiti keadaan percubaan mempengaruhi kualiti keputusan algoritma variatif. Ini bermakna memilih ansatz yang baik adalah penting dalam VQE. Sekali lagi, ini adalah topik yang kaya dan kompleks. Kita tidak akan membincangkan jenis-jenis ansatz yang berbeza atau asal-usulnya di sini. Jika anda berminat untuk mengetahui lebih lanjut tentang litar kuantum berparameter dan ansatz, anda boleh meneroka pelajaran Ansatz dan bentuk variatif daripada kursus Reka bentuk algoritma variatif, yang memberikan penjelasan dan contoh terperinci tentang ansätze.
Oleh kerana kita akan menggunakan Hamiltonian satu-qubit dalam modul ini, kita perlukan litar kuantum berparameter satu-qubit sebagai ansatz. Kita akan melihat tiga jenis ansätze satu-qubit dalam bahagian berikut. Kita akan membandingkannya dan membincangkan pertimbangan utama dalam memilih ansatz.
(Klasik) Pengoptimum: menala halus litar kuantum
Setelah komputer kuantum mengukur tenaga observable daripada ansatz, parameter ansatz dan nilai tenaga dihantar kepada pengoptimum klasik untuk penalaan. Proses pengoptimuman ini dilakukan pada komputer klasik, biasanya menggunakan pakej saintifik tujuan umum seperti SciPy.
Pengoptimum klasik memperlakukan tenaga yang diukur sebagai fungsi kos. Dalam masalah pengoptimuman, fungsi kos (kadangkala dipanggil fungsi objektif) ialah fungsi matematik yang mengukur seberapa "baik" penyelesaian tertentu. Matlamat pengoptimum adalah untuk mencari set parameter yang meminimumkan fungsi kos ini. Dalam konteks mencari tenaga keadaan asas molekul, tenaga itu sendiri berfungsi sebagai fungsi kos — kita ingin mencari parameter untuk litar kuantum kita ("penyelesaian" kita) yang menghasilkan tenaga serendah mungkin. Pengoptimum klasik menggunakan nilai tenaga yang diukur ini (kos) dan menentukan set parameter yang dioptimumkan seterusnya untuk ansatz kuantum. Parameter-parameter yang dikemas kini ini kemudian dihantar kembali ke litar kuantum, dan proses diulang. Dengan setiap lelaran, pengoptimum klasik menyesuaikan parameter untuk cuba mengurangkan tenaga (meminimumkan fungsi kos) sehingga kriteria penumpuan yang ditetapkan dipenuhi, secara ideal memastikan bahawa tenaga terendah yang mungkin (sepadan dengan keadaan asas molekul bagi jarak ikatan dan set asas tersebut) ditemui.
Terdapat banyak strategi pengoptimuman yang disediakan oleh pakej saintifik seperti SciPy. Anda boleh mendapatkan lebih lanjut dalam pelajaran Gelung pengoptimuman bagi kursus Reka bentuk algoritma variatif. Di sini kita akan menggunakan COBYLA (Constrained Optimization BY Linear Approximations), algoritma pengoptimuman yang sesuai untuk landskap tenaga yang rumit. Khususnya, COBYLA tidak cuba mengira kecerunan fungsi yang dikaji; ini dipanggil pengoptimum bebas kecerunan. Bayangkan anda cuba mencari puncak tertinggi dalam banjaran gunung dengan mata tertutup. Oleh kerana anda tidak dapat melihat keseluruhan landskap, anda mengambil langkah kecil ke pelbagai arah, sambil memeriksa sama ada anda naik atau turun. COBYLA berfungsi dengan cara yang serupa — ia bergerak melalui ruang parameter, menguji nilai yang berbeza, secara beransur-ansur memperbaiki keputusan sehingga menemui yang terbaik.
Kini anda bersedia untuk menjalankan pengiraan VQE. Untuk itu, cuba soalan daftar masuk di bawah, yang merumuskan keseluruhan proses.
Semak kefahaman anda
Isikan tempat kosong dengan istilah yang betul untuk melengkapkan ringkasan proses VQE, kemudian klik untuk menyemak jawapan anda.
VQE ialah algoritma kuantum variatif, yang menggabungkan kuasa (1) ________ dan pengkomputeran klasik, digunakan untuk mencari (2) __________ molekul. Proses bermula dengan mendefinisikan (3) __________, yang mewakili jumlah tenaga sistem dan bertindak sebagai observable dalam pengukuran kuantum. Seterusnya, kita menyediakan (4) __________, sebuah litar kuantum dengan parameter boleh laras yang mewakili fungsi gelombang percubaan molekul. Parameter-parameter ini dioptimumkan menggunakan (5) __________, algoritma klasik yang menyesuaikan parameter secara berulang untuk meminimumkan tenaga yang diukur. Dalam perbincangan di atas kita menggunakan pengoptimum (6) __________, yang memperhalusi parameter ansatz tanpa memerlukan pengiraan terbitan. Proses berterusan sehingga kita mencapai (7) __________, bermakna kita telah menemui tenaga terendah yang mungkin bagi molekul.
Bank Kata:
- pengoptimum klasik
- tenaga keadaan asas
- cekap perkakasan
- ansatz
- Hamiltonian molekul
- COBYLA
- pengkomputeran kuantum
- penumpuan
Jawapan
1 → pengkomputeran kuantum
2 → tenaga keadaan asas
3 → Hamiltonian molekul
4 → ansatz
5 → pengoptimum klasik
6 → COBYLA
7 → penumpuan
Kira tenaga keadaan asas atom hidrogen dengan VQE
Sekarang, mari gunakan apa yang telah kita pelajari untuk mengira tenaga keadaan asas atom hidrogen. Sepanjang modul ini, kita akan menggunakan rangka kerja untuk pengkomputeran kuantum yang dikenali sebagai "corak Qiskit", yang membahagikan aliran kerja kepada langkah-langkah berikut:
- Langkah 1: Petakan input klasik kepada masalah kuantum
- Langkah 2: Optimumkan masalah untuk pelaksanaan kuantum
- Langkah 3: Jalankan menggunakan primitif Qiskit Runtime
- Langkah 4: Pasca-pemprosesan dan analisis klasik

Kita akan mengikuti langkah-langkah ini secara umum.
Mari mulakan dengan memuatkan beberapa pakej yang diperlukan, termasuk primitif Qiskit Runtime. Kita juga akan memilih komputer kuantum yang paling tidak sibuk yang tersedia untuk kita.
Terdapat kod di bawah untuk menyimpan kelayakan anda pada penggunaan pertama. Pastikan anda memadam maklumat ini daripada notebook selepas menyimpannya ke persekitaran anda, supaya kelayakan anda tidak dikongsi secara tidak sengaja apabila anda berkongsi notebook. Lihat Sediakan akaun IBM Cloud anda dan Mulakan perkhidmatan dalam persekitaran tidak dipercayai untuk panduan lanjut.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
Sel di bawah akan membolehkan anda bertukar antara menggunakan simulator atau perkakasan sebenar sepanjang notebook. Kami mengesyorkan menjalankannya sekarang:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Langkah 1: Petakan masalah kepada litar dan operator kuantum
Kita memulakan pengiraan VQE dengan mendefinisikan Hamiltonian untuk molekul hidrogen () pada jarak ikatan tertentu. Hamiltonian ini mewakili jumlah tenaga sistem dalam sebutan operator qubit, telah dihasilkan dan dipetakan daripada sistem molekul menggunakan prosedur standard: 1) menggunakan set asas STO-6G (koleksi fungsi matematik tertentu yang digunakan untuk menghampiri orbital elektron), 2) menggunakan pemetaan Jordan-Wigner (teknik untuk menterjemahkan operator fermionik yang menggambarkan elektron kepada operator qubit), dan 3) melakukan pengurangan qubit menggunakan pariti Hamiltonian untuk memudahkan masalah.
Seperti yang telah kita terangkan sebelumnya, tenaga keadaan asas yang dikira sangat bergantung pada pemilihan set asas dan geometri molekul (seperti jarak ikatan). Untuk konfigurasi khusus ini dan selepas transformasi-transformasi ini, Hamiltonian qubit yang terhasil adalah mudah:
Di sini, mewakili operator identiti dan mewakili operator Pauli-Z, bertindak pada satu qubit. Pekali-pekali diterbitkan daripada kamiran yang dikira menggunakan set asas STO-6G pada jarak ikatan tertentu ini dengan transformasi yang betul.
Dengan Hamiltonian ini ditakrifkan, kita kini boleh menggunakan VQE untuk mengira tenaga keadaan asasnya. Adalah berguna untuk membandingkan tenaga keadaan asas yang dikira dengan nilai yang dijangkakan. Untuk satu atom hidrogen (H) yang terpencil, tenaga keadaan asas adalah tepat -0.5 Hartree (tanpa kesan relativistik). Mari kita kira tenaga keadaan asas tepat bagi Hamiltonian qubit khusus kita seperti yang ditakrifkan di atas dan bandingkan dengan nilai yang diketahui yang relevan.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Seterusnya, kita perlukan litar kuantum berparameter, sebuah ansatz, untuk menyediakan fungsi gelombang percubaan bagi keadaan asas. Matlamatnya adalah untuk mencari parameter yang meminimumkan nilai jangkaan tenaga . Pilihan ansatz adalah sangat penting kerana ia menentukan set keadaan kuantum yang mungkin boleh disediakan oleh litar kita. Ansatz yang "baik" ialah yang cukup fleksibel untuk mewakili keadaan yang sangat hampir dengan keadaan asas sebenar Hamiltonian yang kita kaji, tetapi tidak terlalu kompleks sehingga memerlukan terlalu banyak parameter atau litar yang terlalu dalam untuk komputer kuantum semasa.
Di sini, kita akan mencuba tiga ansätze satu-qubit yang berbeza untuk melihat mana yang memberikan "liputan" yang lebih baik terhadap keadaan kuantum yang mungkin boleh dimiliki oleh satu qubit. "Liputan" merujuk kepada julat keadaan kuantum yang boleh dihasilkan oleh litar ansatz dengan mengubah parameternya.
Kita akan menggunakan tiga ansätze berdasarkan kombinasi get putaran satu-qubit yang berbeza:
- Ansatz get putaran 1-paksi: Ansatz ini menggunakan putaran hanya sekitar satu paksi tunggal (). Pada sfera Bloch, ini sepadan dengan bergerak hanya di sepanjang bulatan tertentu. Ini adalah yang paling kurang fleksibel dan merangkumi set keadaan yang terhad.
- Dua ansätze get putaran 2-paksi: Ansätze ini menggabungkan putaran sekitar dua paksi berbeza ( dan ). Ini membolehkan kita mencapai bahagian sfera Bloch yang lebih besar, berbanding dengan putaran satu paksi.
Dengan membandingkan keputusan VQE yang diperoleh dengan tiga ansätze ini, kita dapat melihat bagaimana fleksibiliti dan liputan ruang keadaan ansatz mempengaruhi keupayaan kita untuk mencari tenaga keadaan asas sebenar Hamiltonian yang dipermudahkan. Ansatz yang lebih fleksibel berpotensi untuk mencari penghampiran yang lebih baik, tetapi ia mungkin juga lebih sukar untuk pengoptimum klasik.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Sekarang, mari jana 5000 nombor rawak untuk setiap parameter dan plot taburan keadaan kuantum rawak, yang dijana oleh tiga ansätze dengan parameter rawak ini. Anda boleh fikirkan parameter-parameter ini seperti putaran sekitar paksi berbeza pada permukaan sfera. Untuk melihat taburan keadaan kuantum, kita akan menggunakan Sfera Bloch, sfera tiga dimensi yang menunjukkan keadaan satu qubit. Mana-mana titik pada sfera mewakili keadaan qubit yang mungkin, di mana kutub utara dan selatan adalah seperti "0" dan "1" klasik, tetapi qubit juga boleh berada di mana-mana antaranya, menunjukkan sifat kuantum khas seperti superposisi. Mula-mula, sediakan fungsi yang diperlukan untuk memplot sfera Bloch 3D dan sediakan 5000 parameter rawak.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Mari lihat bagaimana ansatz pertama kita berfungsi.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
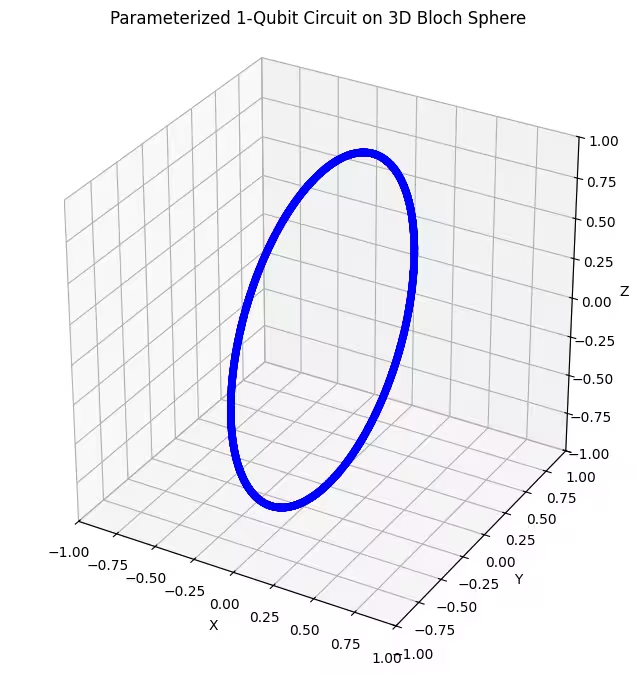
Kita dapat melihat ansatz pertama kita mengembalikan keadaan kuantum yang bertaburan dalam bentuk cincin pada sfera Bloch. Ini masuk akal, kerana kita hanya memberikan ansatz satu parameter putaran. Ia oleh itu hanya boleh menghasilkan keadaan yang diputarkan sekitar satu paksi. Bermula dari titik dan memutar sekitar satu paksi akan sentiasa menghasilkan sebuah cincin. Kemudian mari kita semak ansatz kedua kita, yang mempunyai dua get putaran ortogon - Rx dan Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
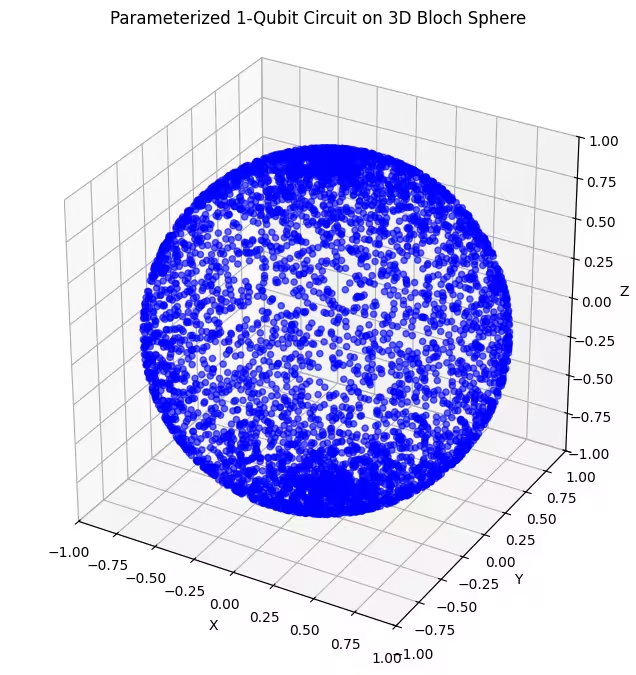
Di sini, kita dapat melihat bahawa ansatz kedua kita merangkumi bahagian yang lebih besar daripada sfera Bloch - tetapi perhatikan bahawa titik-titik lebih tertumpu sekitar kutub dan lebih tersebar sekitar khatulistiwa. Sekarang tiba masanya untuk menyemak ansatz terakhir kita.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
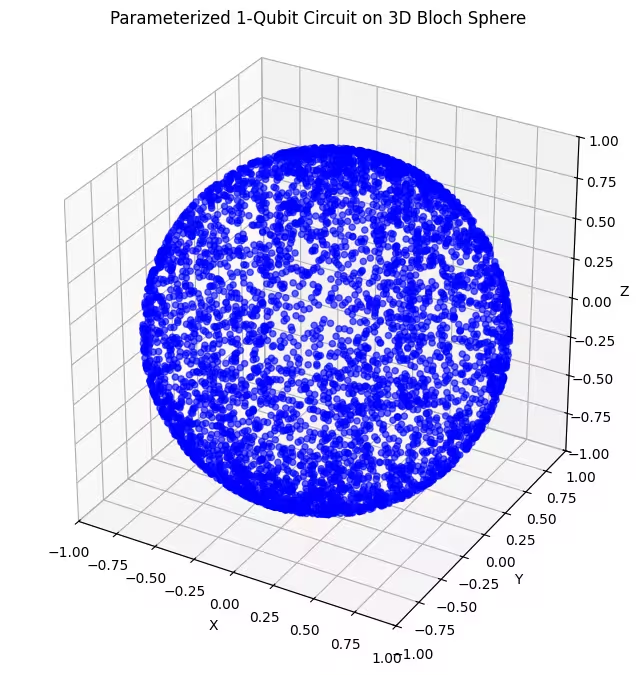
Di sini anda dapat melihat keadaan kuantum yang lebih sekata dihasilkan oleh ansatz terakhir kita.
Seperti yang disebutkan, perkara terbaik yang perlu dilakukan ialah mendapatkan pengetahuan tentang keadaan asas yang anda cari dan menggunakan ansatz yang sesuai untuk meneroka keadaan yang hampir dengan keadaan asas tersebut. Contohnya, jika kita tahu keadaan asas kita adalah berhampiran kutub, kita mungkin memilih ansatz 2. Untuk kesederhanaan, kita akan kekal dengan ansatz 3, yang meneroka keseluruhan sfera Bloch secara sekata.
Sekarang kita telah memilih ansatz kita, mari lukis litar.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Langkah 2: Optimumkan untuk perkakasan sasaran
Apabila menjalankan pengiraan pada komputer kuantum sebenar, kita bukan sahaja mengambil berat tentang logik litar kuantum. Kita juga mengambil berat tentang perkara-perkara seperti operasi apa yang boleh dilakukan oleh komputer kuantum tertentu itu, dan di mana pada komputer kuantum itu qubit yang kita gunakan berada. Adakah mereka bersebelahan antara satu sama lain? Adakah mereka berjauhan? Oleh itu, langkah seterusnya adalah untuk menulis semula litar kita menggunakan get yang semula jadi untuk komputer kuantum yang akan kita gunakan, dan dengan mengambil kira susun atur qubit. Ini boleh dilakukan dengan transpilasi - selepas proses ini, anda dapat melihat ansatz mudah kita ditukar kepada set get yang berbeza, dan qubit abstrak kita akan dipetakan ke dalam qubit fizikal pada komputer kuantum sebenar.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Anda dapat melihat get rx, rz ansatz kita telah ditukar kepada siri get rz, sx, yang merupakan get asli Backend kita. Juga, anda dapat melihat q0 kita kini dipetakan ke dalam qubit fizikal kelima. Kita juga perlu memetakan Hamiltonian kita mengikut perubahan-perubahan ini, seperti dalam kod berikut:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Langkah 3: Jalankan pada perkakasan sasaran
Kini tiba masanya untuk menjalankan VQE kita pada QPU sebenar. Untuk ini, pertama kita perlukan fungsi kos untuk proses pengoptimuman, yang menilai nilai jangkaan Hamiltonian dengan keadaan kuantum, yang dihasilkan oleh ansatz. Jangan risau! Anda tidak perlu mengekod semuanya sendiri. Kami telah menyediakan fungsi untuk ini, dan yang perlu anda lakukan hanyalah menjalankan sel di bawah.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Akhirnya, kita sediakan parameter awal untuk ansatz kita dan proses pengoptimumnya. Anda boleh menggunakan semua sifar atau nilai rawak. Kami telah memilih parameter awal di bawah, tetapi anda bebas untuk mengulas atau menyahulas baris dalam sel untuk mengambil sampel parameter secara rawak, seragam dari 0 hingga .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Tahniah! Anda baru sahaja berjaya menamatkan eksperimen kimia kuantum pertama anda. Kita dapat melihat perbezaan antara tenaga keadaan asas tepat Hamiltonian dan milik kita, tetapi kerana kita menggunakan teknik pengurangan ralat lalai (yang membetulkan ralat bacaan), perbezaannya adalah kecil. Ini adalah permulaan yang sangat baik!
Nota: Anda boleh mendapatkan keputusan yang lebih baik dengan menetapkan tahap pengurangan ralat menggunakan resilience_level. Nilai lalai ialah 1, dan jika anda menetapkan nilai yang lebih tinggi, ia akan menggunakan lebih banyak masa QPU tetapi mungkin mengembalikan keputusan yang lebih baik.
Langkah 4: Pasca-pemprosesan
Tiba masanya untuk melihat bagaimana pengoptimum klasik kita berfungsi. Jalankan sel di bawah dan lihat corak penumpuan.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Kita bermula dengan nilai awal yang agak baik, sehingga kita mendapatkan nilai akhir yang baik hanya dalam 10 langkah. Anda dapat melihat puncak besar dan kecil, dan ini adalah ciri tipikal pengoptimum COBYLA - ia meneroka ruang seolah-olah ia tidak dapat melihat landskap dan menyesuaikan saiz langkah dengan setiap pengukuran.
Semak kefahaman anda
Apakah pemerhatian anda? Bahagian mana daripada proses di atas yang boleh diperbaiki untuk mendapatkan keputusan yang lebih hampir dengan nilai teori, atau lebih hampir dengan tenaga keadaan asas tepat Hamiltonian? Apakah beberapa perkara yang perlu dipertimbangkan untuk ini?
Jawapan
Perkara pertama yang perlu dipertimbangkan ialah perubahan dalam set asas yang digunakan dalam mengira Hamiltonian molekul. Seperti yang disebutkan sebelumnya, tenaga keadaan asas atom H ialah -0.5 Hartree, seperti yang telah diketahui, dan set asas STO-6G yang telah kita pilih tidak mencukupi untuk memperoleh nilai ini secara tepat.
Memilih jenis asas yang lebih kompleks meningkatkan bilangan qubit yang digunakan oleh Hamiltonian; oleh itu, kita perlu memilih ansatz yang lebih kompleks dan sesuai untuk masalah kimia.
Yang seterusnya untuk dioptimumkan ialah pengurusan hingar dalam QPU. Teknik pengurangan ralat yang lebih canggih menghasilkan keputusan yang lebih baik tetapi mungkin mengambil masa yang lebih lama untuk digunakan. Juga, pertimbangkan bagaimana shot_number mempengaruhi keputusan.
Akhirnya, prestasi penumpuan yang lebih baik juga boleh dicapai dengan mencuba pengoptimum yang berbeza.
Kira tenaga keadaan asas molekul hidrogen dengan VQE
Sekarang setelah kita melihat keseluruhan proses VQE menggunakan atom , kita akan mengira tenaga keadaan asas molekul dengan lebih cepat.
Langkah 1: Petakan masalah kepada litar kuantum dan operator
Di sini kami juga menyediakan Hamiltonian satu-qubit yang menggunakan asas STO-6G dan transformasi Jordan-Wigner, dengan pengurangan qubit menggunakan simetri Hamiltonian. Perhatikan bahawa kami menggunakan jarak atom antara dua atom hidrogen sebesar 0.735 .
Tidak seperti pengiraan satu atom hidrogen (), untuk mengira keadaan asas molekul hidrogen (), kita juga perlu mengambil kira daya tolakan antara nukleus dua atom hidrogen, di samping tenaga yang berkaitan dengan orbital elektronik. Dalam langkah ini, kami akan memberikan nilai ini sebagai pemalar, dan kita akan mengira nilai ini secara sebenar dalam soalan semak masuk.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Langkah 2: Optimumkan untuk perkakasan sasaran
Memandangkan bilangan qubit yang digunakan oleh VQE dan Hamiltonian sebelum ini adalah sama dengan backend yang akan digunakan untuk pelaksanaan, kita akan menggunakan ansatz sedia ada beserta bentuk yang telah dioptimumkan.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Langkah 3: Jalankan pada perkakasan sasaran
Kini tiba masanya untuk melakukan pengiraan pada QPU sebenar. Hampir semuanya sama, tetapi kita akan menggunakan titik awal yang sesuai untuk menyesuaikan Hamiltonian. Selain itu, pada bahagian berulang, beberapa tetapan Estimator, yang digunakan untuk mengira jangkaan Hamiltonian bagi ansatz dalam QPU, akan ditetapkan sedikit berbeza daripada pengiraan sebelumnya. Kita akan membincangkan perubahan ini dengan lebih lanjut dalam soalan semak masuk.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Walaupun VQE secara teorinya memberikan batas atas kepada tenaga keadaan asas sebenar, pelaksanaan praktikal pada perkakasan kuantum sebenar atau simulasi berhingar, serta penghampiran yang dibuat dalam menyediakan Hamiltonian (seperti set asas atau pengurangan qubit), boleh memperkenalkan ralat yang kadang-kadang menghasilkan tenaga terukur yang sedikit lebih rendah daripada nilai teori tepat atau nilai rujukan numerik tertentu. Walaupun terdapat beberapa ralat, hasilnya kelihatan memuaskan, terutama memandangkan bilangan langkah yang kecil. Sekarang, mari kita selesaikan pengiraan VQE ini dengan melihat bagaimana pengoptimum berfungsi.
Langkah 4: Pasca-proses
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Semak kefahaman Anda
Mari kita kira tenaga tolakan nukleus molekul , yang kita masukkan sebagai nilai pemalar (0.71997 Hartree).

Gunakan hukum Coulomb dan unit atomik untuk memastikan Anda mendapat nilai dalam Hartree.
Jawapan
Memandangkan kedua-dua nukleus hidrogen bercas positif, mereka saling tolak-menolak disebabkan daya elektrostatik. Tolakan ini diterangkan oleh hukum Coulomb:
di mana ialah cas proton, ialah ketelusan vakum, dan ialah jarak antara dua nukleus, diukur dalam meter atau jejari Bohr dalam unit joule(J).
Untuk mengira tenaga ini dalam Hartree, kita perlu menukar persamaan di atas ke dalam sistem Unit Atomik (AU). Dalam AU, , dan jejari Bohr () adalah 1 dan menjadi skala panjang asas dalam AU. Dengan penyederhanaan ini, hukum Coulomb menjadi:
di mana mesti diukur dalam jejari Bohr ().
Untuk menukar pemisahan nukleus yang diberikan dalam kepada , kita perlukan hubungan penukaran ini:
jadi menjadi .
Oleh itu, tenaga tolakan nukleus bagi yang diberikan ialah
Kira tenaga tindak balas
Sekarang mari gunakan apa yang kita perolehi! Anda telah menggunakan VQE, penyelesai eigen kuantum variasi, untuk mengira tenaga keadaan asas atom dan molekul . Yang tinggal ialah menggunakan nilai yang dikira untuk mendapatkan tenaga tindak balas proses .
Tenaga tindak balas ialah perubahan tenaga yang berlaku apabila bahan-bahan bertindak balas untuk membentuk bahan baru. Bayangkan Anda sedang membina sesuatu: kadang-kadang Anda perlu memasukkan tenaga ke dalamnya (seperti menindih blok), dan kadang-kadang tenaga dilepaskan (seperti bola menggelinding ke bawah bukit). Dalam kimia, tindak balas sama ada menyerap tenaga (endotermik) atau melepaskan tenaga (eksotermik).
Tenaga tindak balas proses boleh dikira dengan formula berikut:
Dengan menjalankan sel di bawah, mari lihat ini secara visual. Di sini kita akan menggunakan nilai keadaan asas tepat bagi setiap Hamiltonian, dan kita akan membandingkan tenaga tindak balas penyelesaian tepat dan hasil VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Seperti yang ditunjukkan dalam rajah, walaupun terdapat beberapa ralat, tenaga keadaan asas tepat Hamiltonian dan tenaga tindak balas yang dikira menggunakan hasil VQE adalah serupa, hampir -0.2 Hartree.
Perlu diperhatikan di sini bahawa tenaga tindak balas proses ini mempunyai nilai negatif, yang bermakna tenaga dilepaskan melalui proses tersebut, dan molekul yang terhasil mempunyai tenaga yang lebih rendah daripada dua atom tunggal. 6. Kesimpulan
Mari kita ringkaskan apa yang telah kita pelajari sejauh ini.
Kita mula-mula melihat dua teknik penghampiran penting yang diperlukan untuk menyelesaikan masalah kimia kuantum: prinsip variasi dan pilihan set asas, yang kedua-duanya adalah asas kepada VQE. Kita meneroka prinsip variasi secara manual, mengira tenaga keadaan asas pengayun harmonik mudah.
Seterusnya, kita meneroka VQE, algoritma yang banyak digunakan untuk mengira tenaga keadaan asas sistem kuantum. Kita menjalankan kod untuk mengira tenaga keadaan asas bagi hidrogen atom () dan molekul hidrogen (). Khususnya, kita belajar bahawa adalah perlu untuk mendapatkan Hamiltonian molekul yang sesuai bagi sistem tersebut dan mengubahnya kepada bentuk yang boleh dilaksanakan pada komputer kuantum. Kita juga melihat bahawa ansatz, litar kuantum berparameter, diperlukan untuk menyediakan keadaan kuantum percubaan dalam VQE, dan kita membincangkan kepentingan memilih struktur litar ansatz yang sesuai. Kita juga belajar bahawa VQE bergantung pada proses pengoptimuman berulang menggunakan komputer klasik, membimbing litar kuantum untuk mencari keadaan tenaga terendah, dan melihat bagaimana proses itu menumpu.
Akhirnya, kita menggunakan tenaga keadaan asas dan yang dikira melalui VQE untuk mengira tenaga tindak balas bagi proses .
VQE adalah algoritma kuantum jangka pendek yang berkuasa, tetapi penting untuk menyedari hadnya. Prestasi VQE sangat bergantung kepada pilihan ansatz – mencari ansatz yang boleh disediakan dengan cekap yang boleh mewakili keadaan asas sebenar dengan tepat menjadi mencabar untuk molekul yang lebih besar dan lebih kompleks. Tambahan pula, perkakasan kuantum semasa mudah terdedah kepada hingar, yang boleh mempengaruhi ketepatan hasil VQE, terutamanya untuk litar yang lebih dalam atau bilangan qubit yang lebih besar. Walaupun menghadapi cabaran ini, VQE berfungsi sebagai algoritma asas, dan penyelidikan berterusan sedang meneroka kaedah variasi yang lebih canggih dan teknik mitigasi ralat untuk menolak sempadan apa yang mungkin dalam kimia kuantum pada komputer kuantum jangka pendek. Sebagai contoh, algoritma seperti Sample-based Quantum Diagonalization (SQD) sedang dibangunkan, yang memanfaatkan sampel yang diperoleh daripada litar kuantum digabungkan dengan pendiagonalan klasik dalam subruang untuk meningkatkan anggaran tenaga dan mengatasi beberapa batasan yang dihadapi oleh VQE, terutamanya berkaitan kecekapan pengukuran dan keteguhan terhadap hingar.
Ulasan dan soalan
Konsep penting:
- Algoritma kuantum variatif ialah paradigma pengkomputeran di mana komputer klasik dan komputer kuantum bekerjasama untuk menyelesaikan sesuatu masalah.
- Dalam VQE, kita bermula dengan Hamiltonian sistem kita dan memetakannya ke atas qubit untuk dilaksanakan pada komputer kuantum. Kita memilih litar kuantum berparameter, iaitu ansatz, dan membuat pengukuran berulang dengan mengubah parameter ansatz, sehingga nilai tenaga terendah dicapai. Pencarian melalui ruang parameter dilakukan menggunakan pengoptimum klasik. Untuk mendapatkan hasil yang baik, perlu memilih ansatz yang baik dan pengoptimum yang sesuai.
- Tenaga tindak balas ialah jumlah perubahan tenaga dalam tindak balas kimia, ditentukan oleh perbezaan antara tenaga reaktan dan produk.
Betul/salah
- Prinsip variatif menyatakan bahawa nilai jangkaan tenaga bagi sebarang fungsi gelombang percubaan sentiasa lebih besar daripada atau sama dengan tenaga keadaan asas sebenar.
- Set asas ialah koleksi fungsi yang digunakan untuk menganggarkan fungsi gelombang kuantum.
- VQE ialah algoritma kuantum yang digunakan untuk menyelesaikan persamaan Schrödinger secara tepat bagi Hamiltonian yang diberikan.
- Dalam VQE, litar kuantum berparameter (ansatz) digunakan untuk menyediakan fungsi gelombang percubaan.
- Pilihan pengoptimum dalam VQE (contohnya, COBYLA, SPSA, atau ADAM) tidak mempengaruhi kualiti keputusan.
EstimatorQiskit digunakan untuk mengira secara langsung nilai jangkaan Hamiltonian dalam VQE.
Soalan aneka pilihan:
- Apakah tujuan Hamiltonian dalam VQE?
- A) Untuk menjana keadaan kuantum rawak
- B) Untuk menentukan tenaga keadaan kuantum
- C) Untuk mengoptimumkan litar kuantum
- D) Untuk mewujudkan keterikatan
- Apakah objektif utama algoritma VQE?
- A) Untuk mencari tenaga keadaan asas suatu Hamiltonian
- B) Untuk mewujudkan keterikatan antara qubit
- C) Untuk melaksanakan carian Grover
- D) Untuk memecahkan penyulitan RSA
- Berapa banyak keadaan kuantum yang dijana dalam buku nota ini untuk membandingkan ansatz?
- A) 100
- B) 1000
- C) 5000
- D) 10,000
- Mengapa pengoptimum klasik diperlukan dalam VQE?
- A) Untuk melaksanakan pengukuran kuantum
- B) Mengemas kini parameter ansatz untuk meminimumkan tenaga
- C) Untuk menyekat qubit
- D) Untuk menjana kerawakan kuantum
- Mengapa ansatz direka untuk berparameter?
- A) Untuk membolehkan penyediaan keadaan kuantum
- B) Untuk membolehkan ruang keadaan kuantum yang luas dicari
- C) Untuk mengurangkan kerumitan litar
- D) Untuk mengukur nilai eigen secara langsung
- Yang manakah pernyataan paling tepat tentang memilih ansatz yang baik?
- A) Ansatz mesti menghasilkan keadaan yang diedarkan secara seragam pada sfera Bloch, atau ia akan gagal.
- B) Ansatz perlu disesuaikan dengan sistem anda untuk memastikan ia boleh menjana keadaan yang hampir dengan keadaan asas.
- C) Ansatz perlu menghasilkan keadaan rawak menggunakan parameter variatifnya.
- D) Ansatz yang lebih baik sentiasa mempunyai lebih banyak parameter variatif.
(Pilihan) Lampiran: Overhed pengoptimum mengikut kerumitan ansatz
VQE menghadapi beberapa cabaran yang sudah diketahui[ref 6], dan berikut adalah yang berkaitan dengan apa yang telah kita pelajari di atas.
- Cabaran pemilihan ansatz
Terdapat cabaran intrinsik dalam memilih ansatz variatif yang tepat. Ansatz berinspirasi kimia (seperti UCCSD) memberikan ketepatan fizikal tetapi memerlukan litar yang dalam, manakala ansatz cekap perkakasan mempunyai litar yang lebih cetek tetapi mungkin kurang interpretasi fizikal. Selain itu, banyak ansatz memperkenalkan parameter variatif berlebihan yang menyumbang sedikit kepada peningkatan ketepatan tetapi meningkatkan kesukaran pengoptimuman dengan ketara.
- Kesukaran pengoptimuman
Landskap pengoptimuman VQE boleh mempunyai kawasan di mana kecerunan hilang secara eksponen (barren plateaus), menjadikannya sukar bagi pengoptimum klasik untuk mengemas kini parameter variatif dengan cekap. Untuk ini, penyelidik telah mencuba menggunakan pelbagai jenis pengoptimum — berasaskan kecerunan dan tanpa kecerunan, tetapi kedua-duanya menghadapi cabaran. Pengoptimum berasaskan kecerunan menderita daripada barren plateaus, manakala kaedah tanpa kecerunan memerlukan bilangan penilaian fungsi yang besar.
- Overhed pengoptimum
Satu lagi cabaran yang sudah diketahui ialah overhed pengoptimum, yang berkaitan dengan skala masalah. litar kuantum yang diperlukan untuk VQE bertambah dalam dan rumit apabila saiz masalah meningkat; ini biasanya juga meningkatkan bilangan parameter untuk dioptimumkan. Proses pengoptimuman menjadi tidak praktikal apabila bilangan parameter meningkat, menyebabkan penumpuan yang perlahan dan kesukaran dalam mencari penyelesaian optimum.
Di sini kita akan melihat cabaran-cabaran ini dengan menggunakan VQE untuk molekul , dengan dua jenis ansatz yang berbeza.
(Nota: Ini boleh mengambil lebih banyak masa QPU, jadi gunakan simulator jika anda tidak mempunyai masa yang cukup.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Sekarang mari jalankan VQE dengan titik awal yang terdiri daripada semua satu, dengan maksimum 20 langkah, dan bandingkan penumpuan kedua-dua jalankan.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Graf di atas dengan jelas menunjukkan bahawa proses pengoptimuman ansatz dengan lebih banyak pemboleh ubah mengambil lebih banyak masa untuk mencapai penumpuan yang stabil.
Berbanding dengan bergantung pada litar qubit tunggal yang mudah dan ansatz yang ringkas, kerumitan pengoptimuman meningkat apabila litar kuantum yang lebih besar dan ansatz berstruktur lebih kompleks diperlukan. Ini menyerlahkan cabaran yang sudah diketahui dalam VQE: overhed pengoptimum.
Penyelidik terus membangunkan pelbagai metodologi canggih yang boleh menggunakan komputer kuantum untuk masalah kimia. Anda boleh mengakses pelbagai bahan pendidikan di IBM Quantum Learning.
Rujukan
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)